- DÉTOXICATION
- DÉTOXICATIONLe terme détoxication recouvre l’ensemble des biotransformations que subissent certains composés exogènes (médicaments par exemple) ou endogènes (hormones stéroïdes, bilirubine) en vue de leur élimination de l’organisme. Ces composés possèdent un caractère hydrophobe, c’est-à-dire qu’ils sont insolubles dans l’eau et solubles dans les lipides. De ce fait, ils ne peuvent être éliminés directement dans les urines, car ils traversent aisément les membranes lipidiques des tubules rénaux et sont donc réabsorbés. Les biotransformations caractéristiques de la détoxication auront une double conséquence: d’une part, rendre plus solubles dans l’eau des structures hydrophobes, et ainsi permettre leur élimination urinaire, accessoirement biliaire; d’autre part, leur faire perdre leurs activités thérapeutiques (médicaments) ou biologiques (hormones). Le terme de détoxication s’est donc écarté de son sens étymologique, puisque le processus porte sur des composés qui ne sont pas, en général, toxiques aux concentrations usuelles, mais qui le deviendraient s’ils s’accumulaient dans l’organisme. Par ailleurs, la définition ci-dessus exclut la «détoxication» de molécules non hydrophobes résultant du métabolisme des nutriments, par exemple celle de l’ammoniaque qui provient du catabolisme protidique et est principalement éliminée sous forme d’urée.Le lieuLes activités de détoxication les plus importantes se situent dans les tissus en interface avec l’environnement: tube digestif, appareil respiratoire, rein, mais surtout foie qui constitue la plaque tournante de ces processus. Au sein de chaque type cellulaire, les principales enzymes de détoxication se localisent dans un système membranaire intracellulaire, le réticulum endoplasmique , où les substrats hydrophobes de ces enzymes tendent à se concentrer. Lorsqu’on souhaite mesurer l’activité de ces enzymes, ou les purifier, on pratique une homogénéisation tissulaire, qui transforme le réticulum endoplasmique en vésicules closes, les microsomes , que l’on peut séparer par ultracentrifugation. À titre d’exemple, les systèmes décrits par les formules 1 et 2, mono-oxygénases à cytochrome P-450 et UDP-glucuronosyltransférases, se situent dans le réticulum endoplasmique, et dans les microsomes après homogénéisation.Les étapesLa détoxication évolue généralement en deux phases: les réactions de phase I représentent des transformations chimiques élémentaires du substrat, oxydation, réduction, hydrolyse par exemple; la phase II consiste en un ensemble de réactions de conjugaison , qui ajoutent au substrat un groupement très polaire favorisant sa solubilité dans l’eau (glucuronoconjugaison, sulfoconjugaison, conjugaison à la glycine, à la glutamine...). Si le composé à détoxiquer possède déjà une fonction chimique permettant l’ancrage de ces groupements polaires, il peut subir directement l’action des enzymes de conjugaison de phase II: c’est le cas de l’acide salicylique, qui est d’emblée glucuronoconjugué. Dans d’autres cas, les réactions de phase I font apparaître des fonctions chimiques permettant ensuite l’action des enzymes de phase II. Soit XH un composé chimique hydrophobe, un médicament par exemple. Une oxygénation (phase I) fait apparaître une fonction 漣OH qui permettra l’ancrage, par une réaction de phase II, du groupe polaire R (de l’acide glucuronique par exemple).Un exemple typique d’un tel schéma de détoxication est fourni par le phénobarbital (ou gardénal, un barbiturique). Il subit tout d’abord l’action d’une enzyme de phase I, une mono-oxygénase à cytochrome P-450, qui fait apparaître une fonction hydroxyle 漣OH; dans un second temps, une enzyme de phase II, l’UDP-glucuronosyltransférase, greffe sur cette fonction un groupement glucuronate (fig. 1).Les métabolites formés sont pharmacologiquement inactifs. Le produit final, le glucuronide de p-hydroxyphénobarbital, très polaire, sera aisément éliminé dans les urines.Les enzymesEnzymes majeures de phase I, les monooxygénases à cytochrome P-450 sont des hémoprotéines (contenant donc du fer), comme l’hémoglobine, mais elles s’en distinguent par leur activité catalytique. En l’absence de substrat, le fer des cytochromes P-450 est à l’état ferrique (Fe III). Après fixation du substrat, deux électrons sont transférés par une réductase à partir d’une coenzyme d’oxydoréduction, le nicotinamide-adénine-dinucléotide phosphate réduit (NADPH, H+). Le premier électron permet le passage du fer à l’état ferreux (Fe II), suivi de la fixation de l’oxygène moléculaire 2 ou dioxygène. Le second électron transforme cet oxygène en espèces réduites beaucoup plus réactives, qui permettent la fixation d’un atome O sur le substrat, d’où le nom de mono-oxygénase . L’autre atome d’oxygène est retrouvé sous forme d’eau H2O. La réaction globale s’écrit (XH représente le substrat hydrophobe de la réaction):
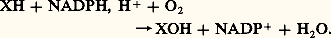 Lorsque (comme c’est le cas décrit ci-dessus) le produit de la réaction possède une fonction hydroxyle 漣OH, alcool ou phénol, la mono-oxygénase est appelée hydroxylase. Dans d’autres cas, la fixation d’oxygène sur une double liaison fait par exemple apparaître un époxyde, et l’enzyme est intitulée époxydase:D’autres types de réactions peuvent être catalysés par des mono-oxygénases, suivant le même schéma général: désalkylations, N-ou S-oxydations...Lorsqu’on réduit chimiquement le cytochrome P-450 et qu’on le soumet à un flux de monoxyde de carbone CO, il développe une bande d’absorption de la lumière très intense à 450 nanomètres: d’où son nom (pigment absorbant à 450 nm). Cette même propriété permet son dosage par spectrophotométrie.La formation de métabolites réactifsNormalement dévolu à la détoxication, le système mono-oxygénasique entraîne à l’occasion des phénomènes d’activation toxique à l’origine de véritables «bavures». En effet, quelques substrats non toxiques par eux-mêmes sont transformés par les mono-oxygénases en métabolites réactifs , doués d’une forte réactivité chimique qui leur permet de se fixer de façon covalente sur des macromolécules cellulaires, protéines ou acides nucléiques. Lorsque ces métabolites réactifs apparaissent en petite quantité, diverses enzymes de phase II (époxyde-hydrolase, glutathion-transférases, UDP-glucuronosyltransférases) les transforment en molécules généralement sans danger. Mais, si leur production devient trop importante, les systèmes de phase II sont débordés et la fixation covalente des métabolites réactifs sur les macromolécules entraîne des effets délétères sur les fonctions cellulaires. À titre d’exemple, le paracétamol est un médicament souvent utilisé à la place de l’aspirine, en particulier dans les pays anglo-saxons. À posologie normale, il ne donne jamais d’accident. Mais une dose trop importante (accidentelle, ou dans un but de suicide) peut entraîner une forte hépatotoxicité (fig. 2).Lorsque les métabolites réactifs se fixent sur l’ADN, ils peuvent entraîner des modifications (mutations somatiques) dont la répétition est susceptible de favoriser l’apparition de cancers. Ainsi l’activation toxique du benzopyrène contenu dans la fumée de tabac a-t-elle été décrite comme un des facteurs possibles de la cancérogenèse pulmonaire. Le benzopyrène est un hydrocarbure aromatique polycyclique sans toxicité directe. Certaines cellules pulmonaires contiennent une mono-oxygénase capable de le transformer en un époxyde qui subit aussitôt l’action de l’époxyde-hydrolase. Le diol ainsi formé peut être repris une seconde fois par une mono-oxygénase à cytochrome P-450, dont l’action produit un diol-époxyde (fig. 3).Ce produit final peut s’intercaler entre les plans de bases de l’ADN et réagir, par son époxyde, avec certaines bases pour former un adduit stable. Au moment de la réplication de l’ADN, cet adduit conduit à de mauvaises reconnaissances de bases complémentaires, et donc à des mutations qui pourraient favoriser l’apparition d’un cancer.D’autres composés chimiques peuvent subir dans l’organisme des transformations analogues les rendant potentiellement cancérigènes: il est possible de les dépister par le test d’Ames , où les métabolites formés par les microsomes hépatiques diffusent vers des micro-organismes en y introduisant des mutations dont on détermine la fréquence par l’utilisation de milieux sélectifs. Le test est actuellement utilisé de façon systématique pour le criblage des médicaments, des additifs alimentaires, des pesticides, des cosmétiques... Il ne donne bien entendu qu’une présomption du caractère cancérigène, d’autres tests, sur animal entier, permettant de le confirmer ou de l’infirmer, au moins dans l’espèce correspondante.Formes multiples et polymorphismeSe trouvant confrontés à des structures chimiques très variables et toujours nouvelles, les enzymes de détoxication peuvent théoriquement faire face à cette situation de deux manières extrêmes: soit une seule enzyme ne reconnaît que le caractère hydrophobe des substrats sans aucune spécificité de structure; soit au contraire il existe un nombre considérable de formes moléculaires multiples d’une même enzyme, chacune spécifique d’une seule structure ou d’un petit nombre de structures proches – système analogue à celui qui régit la reconnaissance antigène-anticorps. La réalité se situe à mi-chemin entre ces deux extrêmes: il existe des familles d’isoenzymes de spécificité assez large et chevauchante, et en nombre probablement limité. C’est le cas, par exemple, des cytochromes P-450, des UDP-glucuronosyltransférases, des glutathion-transférases.À titre d’exemple, les cytochromes P-450 existent, dans chaque espèce, sous des formes moléculaires multiples qui ont pu être regroupées en «familles» et en sousfamilles» caractérisées par leurs analogies de séquences en bases au niveau de l’ADN, en acides aminés au niveau des protéines. Parmi les sous-familles de cytochromes P-450, certaines sont dites constitutives , c’est-à-dire qu’elles existent chez des animaux non traités et présentent une activité particulière, souvent liée au sexe, vis-à-vis de substrats endogènes tels que les hormones stéroïdes. D’autres sont inductibles : elles n’existent pas, ou existent en faible quantité, chez les animaux non traités, mais leur concentration augmente de façon parfois considérable sous l’effet de l’administration, à l’animal, de composés chimiques dits inducteurs. Le tableau présente une classification récente de quelques sous-familles de cytochromes P-450, et, pour certaines d’entre elles, des inducteurs correspondants (cf. tableau).La comparaison des séquences en acides aminés des cytochromes P-450 a montré qu’il existait, au sein d’une même sous-famille, d’importantes analogies de séquence entre formes homologues provenant de différentes espèces animales et de tissus humains. Plusieurs formes de cytochromes P-450 ont maintenant été reconnues chez l’homme, soit par purification directe des protéines à partir de tissus humains, soit par recherche de séquences homologues au niveau du génome ou des ARN messagers à l’aide de «sondes» préparées chez l’animal. L’inductibilité de certaines de ces formes humaines a pu être montrée directement dans quelques cas sur des cultures cellulaires. Elle est plus difficile à prouver in vivo, sinon par des tests indirects de fiabilité variable. Cependant, l’existence de formes moléculaires multiples de mono-oxygénases et l’inductibilité de certaines d’entre elles ont de très importantes conséquences pharmacologiques, notamment dans le cadre des interactions médicamenteuses . Si deux médicaments donnés simultanément sont métabolisés par une même forme moléculaire de monooxygénase, chacun d’entre eux inhibe la biotransformation de l’autre, et leur durée de vie augmente dans l’organisme. Si, de deux médicaments A et B donnés simultanément, l’un (A) est inducteur d’une forme de cytochrome P-450 qui métabolise l’autre (B), la durée de vie de B est au contraire diminuée dans l’organisme. De nombreux exemples d’interactions de ces deux types sont connus: ainsi la cimétidine, utilisée dans le traitement des ulcères gastriques, présente une forte affinité pour certains cytochromes P-450 et inhibe le métabolisme de nombreux autres médicaments, celui de la phénytoïne, un antiépileptique, par exemple; le phénobarbital, puissant inducteur, augmente la vitesse de biotransformation (et donc diminue l’effet) des anticoagulants et des contraceptifs oraux; l’association, en traitement prolongé de la tuberculose, de la rifampicine (un inducteur) à l’isoniazide, augmente les risques d’activation toxique de ce dernier médicament, et son hépatotoxicité potentielle. L’éthanol (ou alcool éthylique) peut agir de deux manières contradictoires, à la fois comme inhibiteur en prise aiguë et comme inducteur de façon chronique, ce qui rend difficile l’ajustement posologique de certains médicaments chez les alcooliques. Enfin, quelques médicaments, de même que l’éthanol, sont susceptibles d’inhiber ou au contraire d’induire la biotransformation de composés endogènes tels que les hormones stéroïdes, d’où d’éventuelles conséquences endocrinologiques de l’alcoolisme chronique, ou de l’utilisation continue du kétoconazole (un antimycotique) par exemple.Dans le cadre des interactions décrites ci-dessus, l’activité des enzymes de détoxication dépend donc de facteurs exogènes; mais elle est aussi modulée par des facteurs endogènes, constitutionnels ou acquis, par exemple l’âge, le sexe ou une situation pathologique telle que le diabète; elle est également sous contrôle génétique: ce contrôle et ses conséquences pharmacologiques sont étudiés par la pharmacogénétique . Certains polymorphismes d’activité des enzymes de détoxication, anciennement connus, concernent le métabolisme des curarisants (polymorphisme de la pseudocholinestérase) ou de l’isoniazide («acétyleurs rapides» ou «acétyleurs lents» suivant l’activité de l’acétyltransférase, enzyme de phase II). Des polymorphismes génétiques d’oxydation, atteignant certaines formes moléculaires de cytochrome P-450, ont été plus récemment identifiés. L’un des mieux étudiés touche une mono-oxygénase active sur des antihypertenseurs (débrisoquine, bufuralol), des tranquillisants et d’autres médicaments. Moins de 10 p. 100 de la population européenne ou nord-américaine présente une activité très faible de cette mono-oxygénase: chez ces «hydroxylateurs lents» existent, à posologie égale, plus de risques d’accumulation des médicaments métabolisés par cette forme que chez les «hydroxylateurs rapides». Les polymorphismes d’activité basale, ou d’inductibilité, de formes d’enzymes impliquées dans le processus d’activation de composés cancérigènes pourraient influencer la prédisposition génétique à certains cancers. Ce serait le cas par exemple du polymorphisme «débrisoquine-bufuralol» décrit ci-dessus. Quelques groupes ont également corrélé l’inductibilité de l’«arylhydrocarbon hydroxylase », activité portée par les cytochromes P-450IA, dans les lymphocytes humains cultivés in vitro, aux risques d’apparition du cancer du poumon chez les fumeurs.
Lorsque (comme c’est le cas décrit ci-dessus) le produit de la réaction possède une fonction hydroxyle 漣OH, alcool ou phénol, la mono-oxygénase est appelée hydroxylase. Dans d’autres cas, la fixation d’oxygène sur une double liaison fait par exemple apparaître un époxyde, et l’enzyme est intitulée époxydase:D’autres types de réactions peuvent être catalysés par des mono-oxygénases, suivant le même schéma général: désalkylations, N-ou S-oxydations...Lorsqu’on réduit chimiquement le cytochrome P-450 et qu’on le soumet à un flux de monoxyde de carbone CO, il développe une bande d’absorption de la lumière très intense à 450 nanomètres: d’où son nom (pigment absorbant à 450 nm). Cette même propriété permet son dosage par spectrophotométrie.La formation de métabolites réactifsNormalement dévolu à la détoxication, le système mono-oxygénasique entraîne à l’occasion des phénomènes d’activation toxique à l’origine de véritables «bavures». En effet, quelques substrats non toxiques par eux-mêmes sont transformés par les mono-oxygénases en métabolites réactifs , doués d’une forte réactivité chimique qui leur permet de se fixer de façon covalente sur des macromolécules cellulaires, protéines ou acides nucléiques. Lorsque ces métabolites réactifs apparaissent en petite quantité, diverses enzymes de phase II (époxyde-hydrolase, glutathion-transférases, UDP-glucuronosyltransférases) les transforment en molécules généralement sans danger. Mais, si leur production devient trop importante, les systèmes de phase II sont débordés et la fixation covalente des métabolites réactifs sur les macromolécules entraîne des effets délétères sur les fonctions cellulaires. À titre d’exemple, le paracétamol est un médicament souvent utilisé à la place de l’aspirine, en particulier dans les pays anglo-saxons. À posologie normale, il ne donne jamais d’accident. Mais une dose trop importante (accidentelle, ou dans un but de suicide) peut entraîner une forte hépatotoxicité (fig. 2).Lorsque les métabolites réactifs se fixent sur l’ADN, ils peuvent entraîner des modifications (mutations somatiques) dont la répétition est susceptible de favoriser l’apparition de cancers. Ainsi l’activation toxique du benzopyrène contenu dans la fumée de tabac a-t-elle été décrite comme un des facteurs possibles de la cancérogenèse pulmonaire. Le benzopyrène est un hydrocarbure aromatique polycyclique sans toxicité directe. Certaines cellules pulmonaires contiennent une mono-oxygénase capable de le transformer en un époxyde qui subit aussitôt l’action de l’époxyde-hydrolase. Le diol ainsi formé peut être repris une seconde fois par une mono-oxygénase à cytochrome P-450, dont l’action produit un diol-époxyde (fig. 3).Ce produit final peut s’intercaler entre les plans de bases de l’ADN et réagir, par son époxyde, avec certaines bases pour former un adduit stable. Au moment de la réplication de l’ADN, cet adduit conduit à de mauvaises reconnaissances de bases complémentaires, et donc à des mutations qui pourraient favoriser l’apparition d’un cancer.D’autres composés chimiques peuvent subir dans l’organisme des transformations analogues les rendant potentiellement cancérigènes: il est possible de les dépister par le test d’Ames , où les métabolites formés par les microsomes hépatiques diffusent vers des micro-organismes en y introduisant des mutations dont on détermine la fréquence par l’utilisation de milieux sélectifs. Le test est actuellement utilisé de façon systématique pour le criblage des médicaments, des additifs alimentaires, des pesticides, des cosmétiques... Il ne donne bien entendu qu’une présomption du caractère cancérigène, d’autres tests, sur animal entier, permettant de le confirmer ou de l’infirmer, au moins dans l’espèce correspondante.Formes multiples et polymorphismeSe trouvant confrontés à des structures chimiques très variables et toujours nouvelles, les enzymes de détoxication peuvent théoriquement faire face à cette situation de deux manières extrêmes: soit une seule enzyme ne reconnaît que le caractère hydrophobe des substrats sans aucune spécificité de structure; soit au contraire il existe un nombre considérable de formes moléculaires multiples d’une même enzyme, chacune spécifique d’une seule structure ou d’un petit nombre de structures proches – système analogue à celui qui régit la reconnaissance antigène-anticorps. La réalité se situe à mi-chemin entre ces deux extrêmes: il existe des familles d’isoenzymes de spécificité assez large et chevauchante, et en nombre probablement limité. C’est le cas, par exemple, des cytochromes P-450, des UDP-glucuronosyltransférases, des glutathion-transférases.À titre d’exemple, les cytochromes P-450 existent, dans chaque espèce, sous des formes moléculaires multiples qui ont pu être regroupées en «familles» et en sousfamilles» caractérisées par leurs analogies de séquences en bases au niveau de l’ADN, en acides aminés au niveau des protéines. Parmi les sous-familles de cytochromes P-450, certaines sont dites constitutives , c’est-à-dire qu’elles existent chez des animaux non traités et présentent une activité particulière, souvent liée au sexe, vis-à-vis de substrats endogènes tels que les hormones stéroïdes. D’autres sont inductibles : elles n’existent pas, ou existent en faible quantité, chez les animaux non traités, mais leur concentration augmente de façon parfois considérable sous l’effet de l’administration, à l’animal, de composés chimiques dits inducteurs. Le tableau présente une classification récente de quelques sous-familles de cytochromes P-450, et, pour certaines d’entre elles, des inducteurs correspondants (cf. tableau).La comparaison des séquences en acides aminés des cytochromes P-450 a montré qu’il existait, au sein d’une même sous-famille, d’importantes analogies de séquence entre formes homologues provenant de différentes espèces animales et de tissus humains. Plusieurs formes de cytochromes P-450 ont maintenant été reconnues chez l’homme, soit par purification directe des protéines à partir de tissus humains, soit par recherche de séquences homologues au niveau du génome ou des ARN messagers à l’aide de «sondes» préparées chez l’animal. L’inductibilité de certaines de ces formes humaines a pu être montrée directement dans quelques cas sur des cultures cellulaires. Elle est plus difficile à prouver in vivo, sinon par des tests indirects de fiabilité variable. Cependant, l’existence de formes moléculaires multiples de mono-oxygénases et l’inductibilité de certaines d’entre elles ont de très importantes conséquences pharmacologiques, notamment dans le cadre des interactions médicamenteuses . Si deux médicaments donnés simultanément sont métabolisés par une même forme moléculaire de monooxygénase, chacun d’entre eux inhibe la biotransformation de l’autre, et leur durée de vie augmente dans l’organisme. Si, de deux médicaments A et B donnés simultanément, l’un (A) est inducteur d’une forme de cytochrome P-450 qui métabolise l’autre (B), la durée de vie de B est au contraire diminuée dans l’organisme. De nombreux exemples d’interactions de ces deux types sont connus: ainsi la cimétidine, utilisée dans le traitement des ulcères gastriques, présente une forte affinité pour certains cytochromes P-450 et inhibe le métabolisme de nombreux autres médicaments, celui de la phénytoïne, un antiépileptique, par exemple; le phénobarbital, puissant inducteur, augmente la vitesse de biotransformation (et donc diminue l’effet) des anticoagulants et des contraceptifs oraux; l’association, en traitement prolongé de la tuberculose, de la rifampicine (un inducteur) à l’isoniazide, augmente les risques d’activation toxique de ce dernier médicament, et son hépatotoxicité potentielle. L’éthanol (ou alcool éthylique) peut agir de deux manières contradictoires, à la fois comme inhibiteur en prise aiguë et comme inducteur de façon chronique, ce qui rend difficile l’ajustement posologique de certains médicaments chez les alcooliques. Enfin, quelques médicaments, de même que l’éthanol, sont susceptibles d’inhiber ou au contraire d’induire la biotransformation de composés endogènes tels que les hormones stéroïdes, d’où d’éventuelles conséquences endocrinologiques de l’alcoolisme chronique, ou de l’utilisation continue du kétoconazole (un antimycotique) par exemple.Dans le cadre des interactions décrites ci-dessus, l’activité des enzymes de détoxication dépend donc de facteurs exogènes; mais elle est aussi modulée par des facteurs endogènes, constitutionnels ou acquis, par exemple l’âge, le sexe ou une situation pathologique telle que le diabète; elle est également sous contrôle génétique: ce contrôle et ses conséquences pharmacologiques sont étudiés par la pharmacogénétique . Certains polymorphismes d’activité des enzymes de détoxication, anciennement connus, concernent le métabolisme des curarisants (polymorphisme de la pseudocholinestérase) ou de l’isoniazide («acétyleurs rapides» ou «acétyleurs lents» suivant l’activité de l’acétyltransférase, enzyme de phase II). Des polymorphismes génétiques d’oxydation, atteignant certaines formes moléculaires de cytochrome P-450, ont été plus récemment identifiés. L’un des mieux étudiés touche une mono-oxygénase active sur des antihypertenseurs (débrisoquine, bufuralol), des tranquillisants et d’autres médicaments. Moins de 10 p. 100 de la population européenne ou nord-américaine présente une activité très faible de cette mono-oxygénase: chez ces «hydroxylateurs lents» existent, à posologie égale, plus de risques d’accumulation des médicaments métabolisés par cette forme que chez les «hydroxylateurs rapides». Les polymorphismes d’activité basale, ou d’inductibilité, de formes d’enzymes impliquées dans le processus d’activation de composés cancérigènes pourraient influencer la prédisposition génétique à certains cancers. Ce serait le cas par exemple du polymorphisme «débrisoquine-bufuralol» décrit ci-dessus. Quelques groupes ont également corrélé l’inductibilité de l’«arylhydrocarbon hydroxylase », activité portée par les cytochromes P-450IA, dans les lymphocytes humains cultivés in vitro, aux risques d’apparition du cancer du poumon chez les fumeurs.
détoxication [ detɔksikasjɔ̃ ] n. f.• 1945; de dé- et (in)toxication♦ Physiol. Action de détoxiquer; son résultat.♢ Élimination des toxines par un organisme.
● détoxication nom féminin Processus par lequel l'organisme inactive les substances toxiques d'origine interne ou externe. Élimination du caractère toxique d'un déchet avant traitement ou mise en décharge. (Elle peut consister à neutraliser les déchets acides ou basiques, les uns par les autres ou à l'aide de réactifs ; à modifier l'état d'oxydation de certains ions ; ou à enfermer les ions toxiques dans un réseau siliceux qui les rend insolubles.)détoxicationn. f. MED Neutralisation du pouvoir toxique (de certains corps).— élimination des toxines.détoxication [detɔksikɑsjɔ̃] n. f.ÉTYM. 1945, Garnier et Delamare, in D. D. L.; de 1. dé-, toxique, et suff. -ation, d'après intoxication.❖♦ Physiol. Action de détoxiquer; son résultat. — Élimination des toxines par un organisme.REM. On trouve aussi la forme détoxification (mil. XXe).
Encyclopédie Universelle. 2012.